New Achievements in the Field of Artificial Intelligence by NKU Published on Nature Methods
On March 19, Zhang Han’s team from the College of Artificial Intelligence, Nankai University, in collaboration with Yao Jianhua’s team from Tencent AI Lab, published a paper entitled “scPROTEIN: a versatile deep graph contrastive learning framework for single-cell proteomics embedding” in Nature Methods, a top international academic journal. This study proposed a single-cell proteomics representation learning method called scPROTEIN, which is the first to develop a unified deep learning framework to solve this set of tangled problems about data missingness, batch effects and high noise caused by mass spectrometry sequencing in data processing, and learn accurate cell embedding representation for downstream analyses.
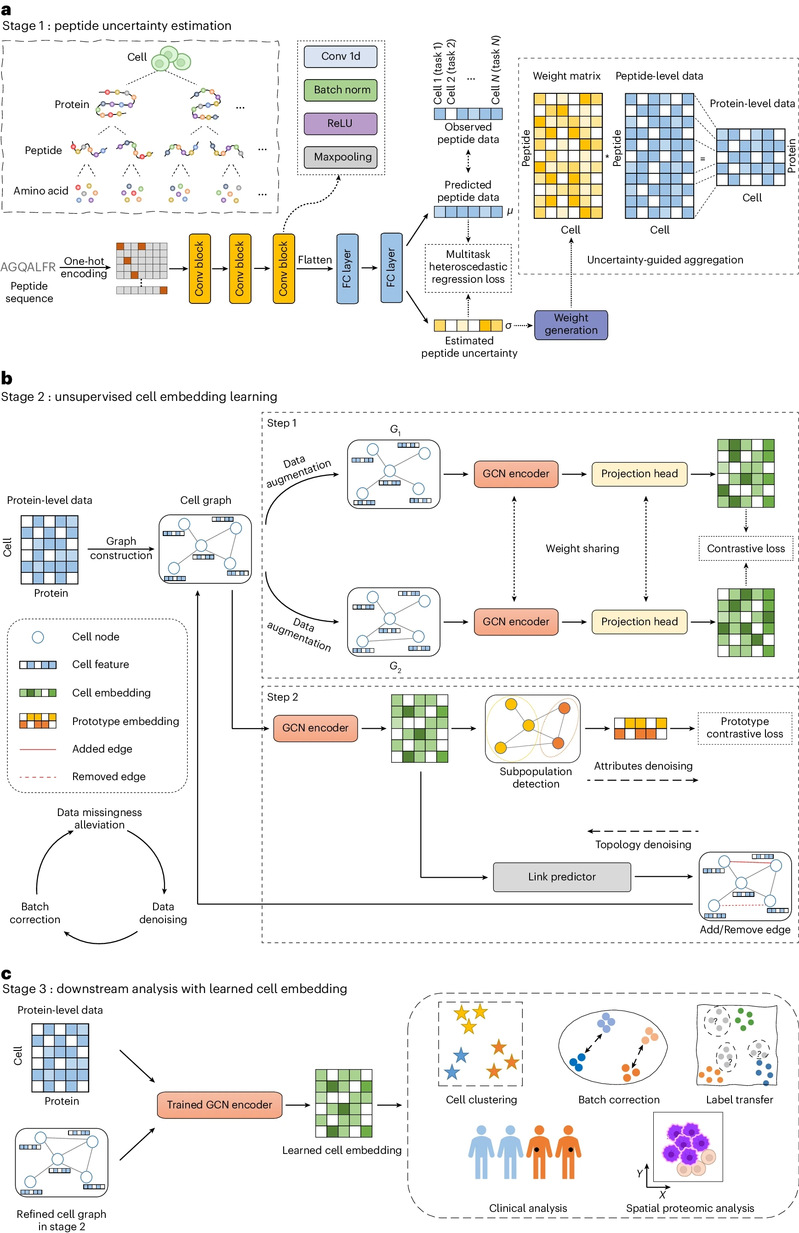
Schematic diagram of the scPROTEIN model
scPROTEIN is a computational method for single-cell proteomics data. Firstly, a multitask heteroscedastic regression model was used to assign uncertainty weights to each peptide signal in each cell, and uncertainty-guided aggregation was designed to obtain protein-level abundance from peptide-level data. It fully utilized the hierarchical structure of proteomics to improve the data quality. scPROTEIN represents single-cell proteomic data by establishing a cell graph structure, and the messaging process on the graph takes full account of the co-expression pattern to alleviate the problem of data missing. The model performs self-supervised training based on graph contrastive learning, and the discriminative nature of contrastive learning can implicitly remove batch effects without relying on data priori knowledge. At the same time, scPROTEIN designs an alternating topology-attribute denoising module to denoise proteomic data and obtain accurate cell representation. Attribute noise reduction, which is based on prototype contrastive learning, utilizes cell prototypes that are far away from the boundaries to enhance information about other cells. The enhanced cell representation is used to dynamically improve the cell map topological structure and helps to obtain more accurate cell representation.
Through comprehensive experiments on downstream tasks, the paper systematically demonstrates the applicability and superior performance of scPROTEIN in mass spectrometry-based and antibody-based proteomics. Compared to existing processing flow for single-cell proteomics data and other transcriptome comparison methods, scPROTEIN has demonstrated better performance in cell clustering, batch correction, and cell type annotation tasks. Furthermore, scPROTEIN has exhibited wide-ranging applicability in both single-cell clinical proteome analysis and protein data analysis with spatial resolution. Following the rapid development and application of single-cell proteomics technology, scPROTEIN will play an increasingly important role in single-cell proteomics data analysis scenarios, providing new methods and tools for interpreting complex biological data. This leading achievement provides a new idea for AI for Science, and demonstrates the tremendous potential of deep learning to solve complex problems in biomedical data analysis.
Nankai University is the first author affiliation. Li Wei, a doctoral student from Nankai University, is the first author. Professor Zhang Han from the College of Artificial Intelligence of Nankai University and Dr. Yao Jianhua from Tencent AI Lab are the co-corresponding authors. Dr. Yang Fan from Tencent AI Lab is the co-first author.
Paper URL:
https://www.nature.com/articles/s41592-024-02214-9
(Edited and translated by Nankai News Team.)
38 Tongyan Road, Jinnan District, Tianjin, P.R.China 300350 94 Weijin Road, Nankai District, Tianjin, P.R.China 300071
Copyright: Nankai University All Rights Reserved






