Nankai University Team Successfully Develops Highly Accurate Protein Structure Prediction Algorithm
An important research result entitled “Deep-learning-based single-domain and multidomain protein structure prediction with D-I-TASSER” was published in Nature Biotechnology on May 23 with Professor Zheng Wei from the School of Statistics and Data Science of Nankai University as the first author.

This study developed D-I-TASSER, a protein structure prediction algorithm that integrates deep learning spatial constraints and statistical energy functions, to achieve highly accurate protein structure prediction, surpassing the performance of the AlphaFold3 algorithm. The corresponding authors of the paper are Professor Zhang Yang of the National University of Singapore and Associate Professor Lydia Freddolino of the University of Michigan.

Workflow of the D-I-TASSER algorithm
D-I-TASSER participated in the 15th CASP (Critical Assessment of Protein Structure Prediction) competition. It ranked first in the world in both single-domain protein and multi-domain protein categories by beating more than 80 academic and industrial research groups around the world, including AlphaFold2 and AlphaFold3. This demonstrates its world-leading position in the prediction of complex protein structures.
D-I-TASSER also performed structural and functional prediction (Gene Ontology term, Enzyme Commission number, and Ligand Binding residues) of proteins in the human proteome. It successfully predicted 80.5% of domain-level protein structures and 72.8% of full-chain protein structures, and accurately predicted 3,020 proteins that were difficult to predict using AlphaFold2, demonstrating its value in compensating the blind spot of structural prediction.
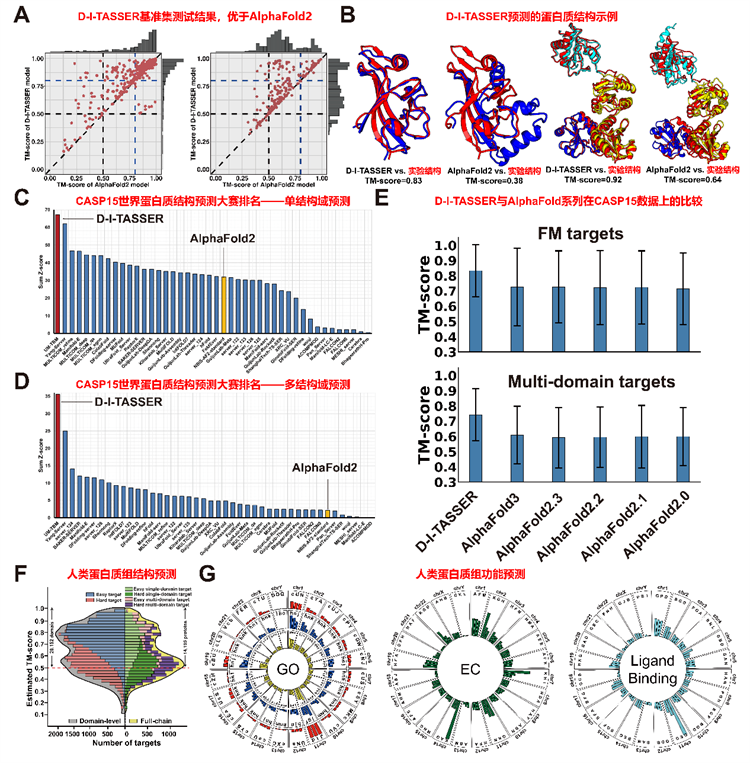
D-I-TASSER performed well in benchmark tests and CASP15 and was successfully applied to predict the structure and functions of the human whole proteome
D-I-TASSER shows good performance in large-scale protein structure prediction, especially in structurally complex and difficult proteins and multi-domain protein predictions, demonstrating a new paradigm that combines deep learning spatial constraints and statistical energy functions. Not only does this method help to understand protein folding and function, but it also results in preliminary progress in antibody screening and optimization, identification of pathogenic genes for rare diseases, prediction of viral infectivity, and structural analysis of auxiliary cryo-EM. The technology is expected to be commercialized through cooperation with relevant research institutions and biomedical enterprises.
Paper link: https://www.nature.com/articles/s41587-025-02654-4
(Edited and translated by Nankai News Team.)
38 Tongyan Road, Jinnan District, Tianjin, P.R.China 300350 94 Weijin Road, Nankai District, Tianjin, P.R.China 300071
Copyright: Nankai University All Rights Reserved






